Noções básicas da regulamentação governamental de dispositivos médicos
(Nota: os textos em nosso espaço de “recursos” estão sempre sendo aprimorados para trazer as melhores explicações sobre tópicos regulatórios e normativos, portanto, salve o link e volte sempre!)
A segurança e desempenho de dispositivos médico depende de dois elementos críticos:
- O produto (O dispositivo médico em si)
- O uso do dispositivo médico
Do ponto de vista de regulamentação, as atividade de revisão pré-mercado (ou pré-comercialização) contribuem para o controle do produto, e a vigilância pós-mercado (ou pós-comercialização) garante que os dispositivos médicos em uso continuem seguros e eficazes.
Um terceiro elemento muito importante é a chamada “representação do produto para o usuário“. Este elemento está relacionado ao fato de que, embora o projeto de um dispositivo médicos possa ter milhares de informações, apenas certas informações, que indicam a maneira segura de usar o dispositivo, chegam ao usuário/ paciente. Isto faz com que esta representação seja essencial para o uso seguro e eficaz.
Ciclo de vida de dispositivos médicos
A figura abaixo mostras as fases gerais do ciclo de vida de dispositivos médicos. Quaisquer dessas fases podem afetar a segurança e eficácia dos dispositivos.
Os estágio do controle regulatório governamental de dispositivos médicos também se baseiam nestas fases.
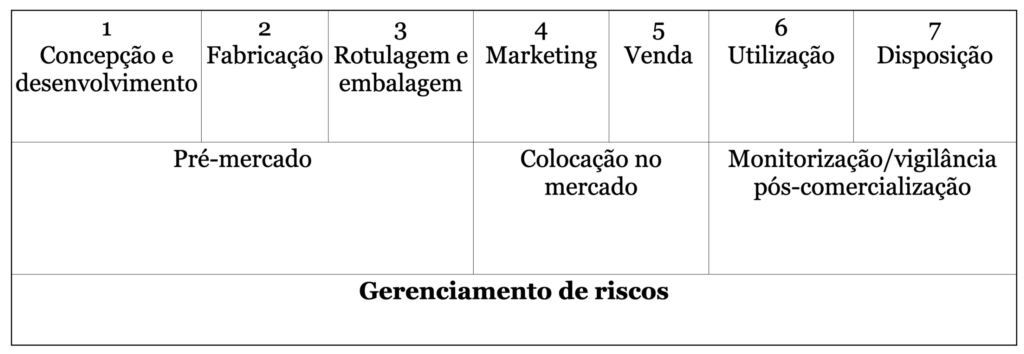
Os controles pré-mercado garantem que o produto a ser colocado no mercado está em conformidade com os requisitos regulatórios aplicáveis.
Controles de rotulagem (termo geral usado para identificar informações fornecidas pelo fabricante para o uso seguro do dispositivo, como o rótulo e instruções de uso) e de publicidade garantem a correta representação do produto para o usuário.
Controle relacionados à vigilância pós-mercado garantem segurança e eficácia continuadas do uso do dispositivo.